
Usher Syndrome and Retinitis Pigmentosa: What Optometrists Should Know
Read about Usher Syndrome in optometry—its causes, types, symptoms, diagnosis, and management. Learn how optometrists play a key role in identifying and supporting patients with this rare genetic disorder.
In the world of eye care, some conditions extend beyond vision alone. Usher Syndrome is one such disorder that affects both hearing and sight, making it a critical area of awareness for optometrists. It is the most common cause of combined congenital deafness and progressive blindness, and though rare, its impact on quality of life is profound.
Understanding Usher Syndrome from an optometric perspective helps clinicians identify early signs of retinitis pigmentosa (RP), offer timely low vision care, and coordinate multidisciplinary management for better patient outcomes.
What is Usher Syndrome?
Usher Syndrome is an autosomal recessive genetic disorder characterized by sensorineural hearing loss and progressive retinal degeneration due to retinitis pigmentosa (RP). Some patients also present with balance (vestibular) dysfunction.
Because the disease affects both the auditory and visual systems, early detection—often by optometrists—is vital for ensuring patients receive appropriate hearing and vision support.
Types of Usher Syndrome
Usher Syndrome is classified into three main types based on the severity and onset of symptoms:
| Type | Hearing Loss | Vision Loss (RP onset) | Balance |
|---|---|---|---|
| Type I | Profound, present at birth | Early childhood | Affected |
| Type II | Moderate, stable | Adolescence | Normal |
| Type III | Progressive, develops later | Teens to adulthood | Variable |
Clinical Features Relevant to Optometry
Optometrists often encounter the visual symptoms of Usher Syndrome before a systemic diagnosis is made. Typical findings include:
- Night blindness (nyctalopia) – often the first symptom
- Progressive peripheral vision loss leading to tunnel vision
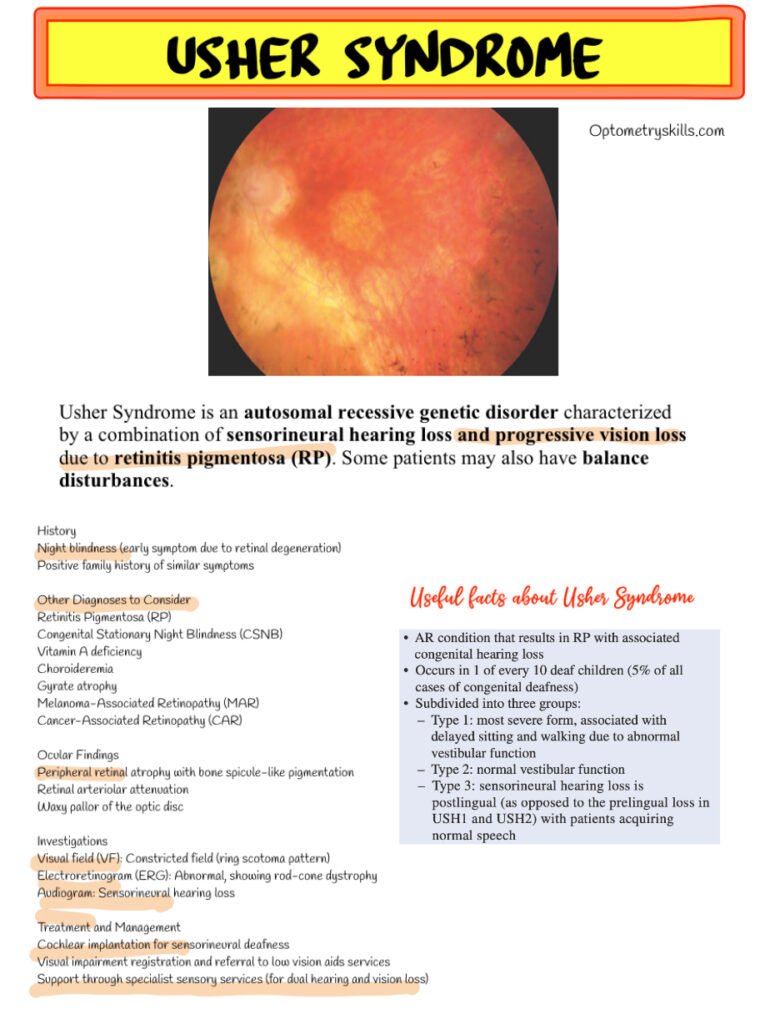
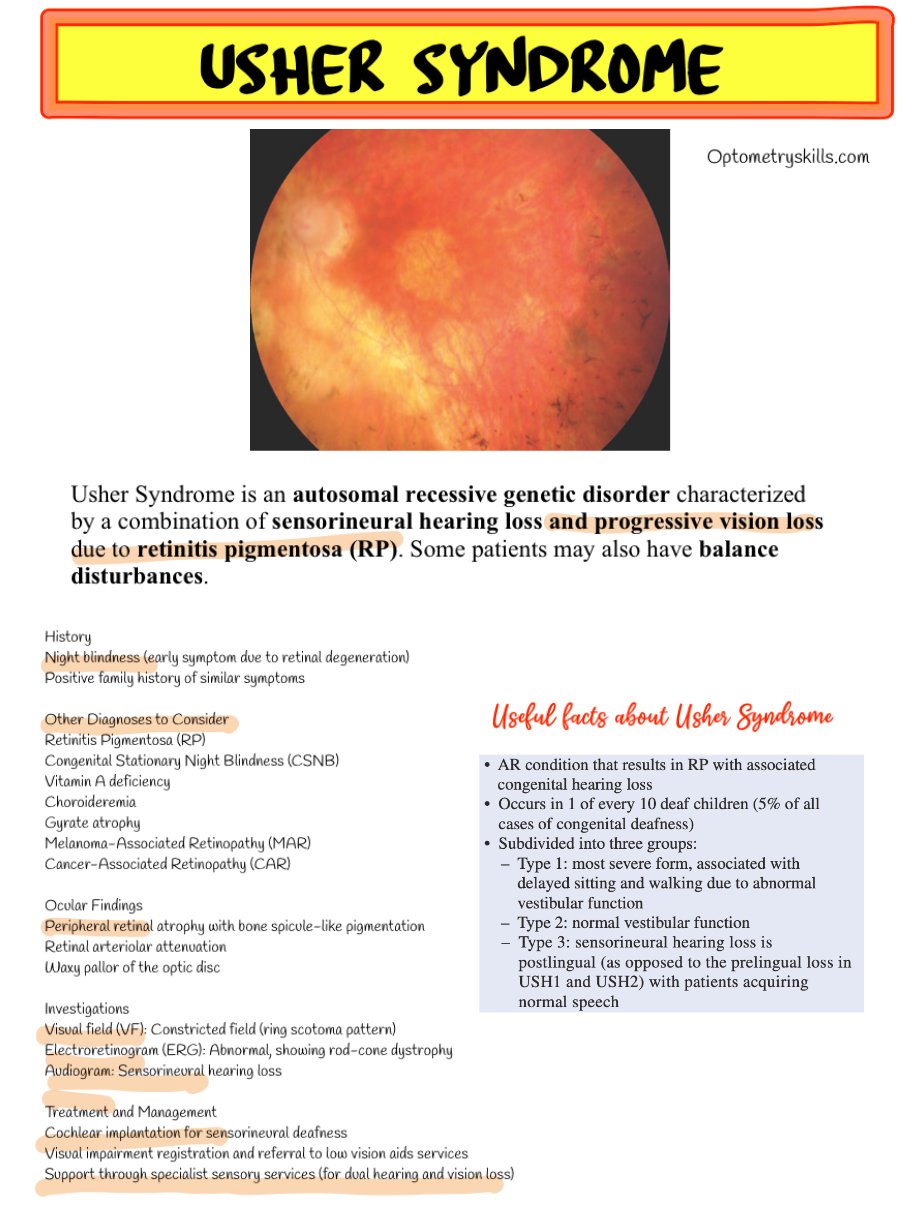
- Bone spicule-like pigmentation in the peripheral retina
- Retinal arteriolar attenuation
- Waxy pallor of the optic disc
These findings are characteristic of retinitis pigmentosa, and optometrists play a key role in identifying them during routine dilated fundus examinations.
Differential Diagnoses
When encountering RP-like retinal changes, consider other possible causes:
- Congenital Stationary Night Blindness (CSNB)
- Vitamin A deficiency
- Choroideremia
- Gyrate Atrophy
- Melanoma-Associated Retinopathy (MAR)
- Cancer-Associated Retinopathy (CAR)
Investigations
A complete evaluation should include both ocular and systemic investigations:
- Visual Field (VF) Testing: Typically shows ring scotomas or constricted fields
- Electroretinogram (ERG): Reveals rod-cone dystrophy pattern
- Audiogram: Detects sensorineural hearing loss
- Genetic Testing: Confirms the diagnosis and type of Usher Syndrome
Role of the Optometrist in Management
While there is no cure for Usher Syndrome, optometrists can make a significant impact through early identification and low vision support. Management includes:
- Referral for cochlear implantation (for hearing rehabilitation)
- Low vision aids: magnifiers, CCTV readers, and adaptive lighting
- Referral to sensory impairment services and rehabilitation centers
- Collaboration with audiologists and genetic counselors
- Patient education about mobility training and lifestyle adaptation
Optometrists can also refer patients to Sense Usher Service and Royal National Institute of Blind People (RNIB) for specialized support and community resources.
Recent Advances in Research
Research into gene therapy and retinal implants offers hope for the future. Clinical trials are exploring gene correction techniques to slow retinal degeneration in Usher Syndrome, particularly in USH2A and MYO7A gene mutations.
Frequently Asked Questions (FAQ)
Q1. What causes Usher Syndrome?
Usher Syndrome is caused by mutations in specific genes responsible for the normal development of inner ear hair cells and retinal photoreceptors. It follows an autosomal recessive inheritance pattern, meaning both parents must carry the faulty gene.
Q2. How does an optometrist detect Usher Syndrome?
Through fundus examination showing RP-like changes, visual field tests indicating peripheral loss, and patient history suggesting night blindness or family history of vision and hearing issues.
Q3. Can Usher Syndrome be cured?
Currently, there is no cure, but management focuses on maximizing remaining vision and hearing through assistive devices and early intervention.
Q4. What age does vision loss start in Usher Syndrome?
It varies by type—early childhood in Type I, adolescence in Type II, and adulthood in Type III.
Q5. How can low vision aids help?
Low vision aids like magnifiers, electronic readers, and orientation training can significantly improve independence and daily functioning.
Conclusion
For optometrists, understanding Usher Syndrome extends beyond diagnosing retinal changes—it’s about recognizing a systemic disorder that requires compassionate, multidisciplinary care. Early identification, timely referral, and vision rehabilitation can help patients maintain independence and improve their quality of life despite the challenges of combined sensory loss.
Follow us in Facebook