
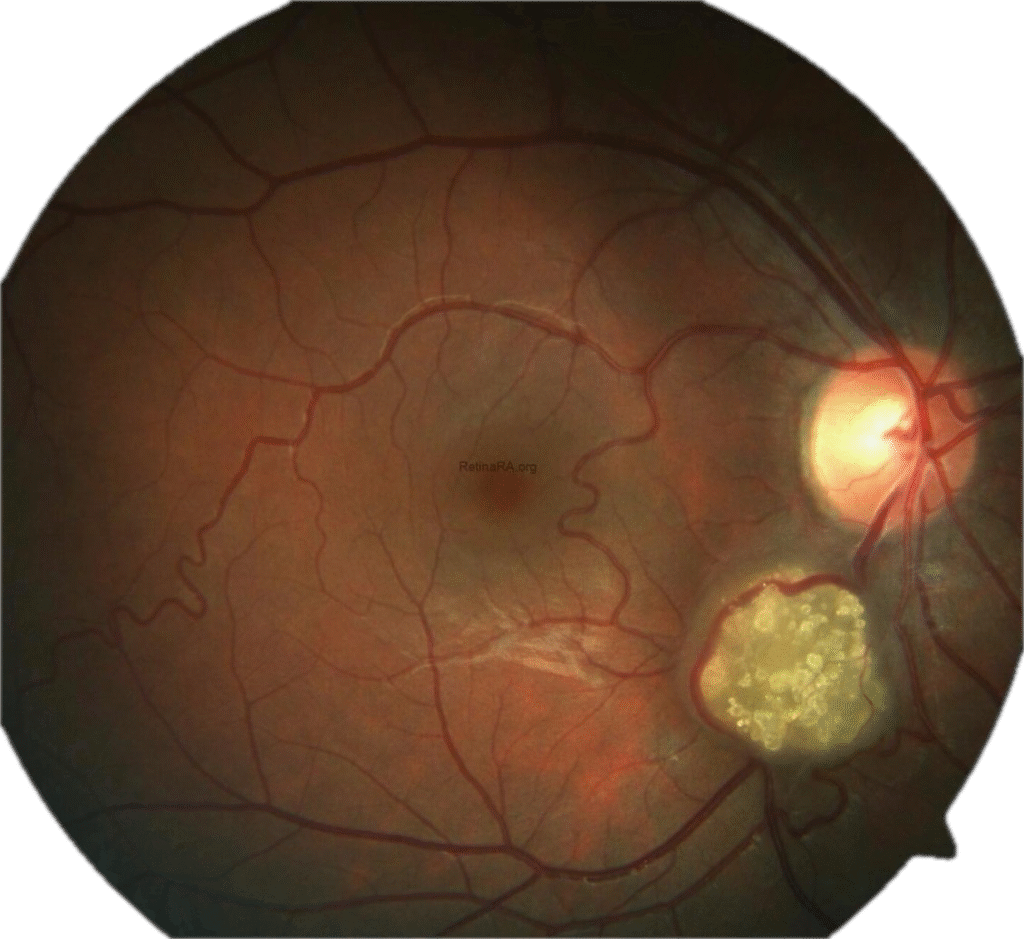
Retinal Astrocytoma (Retinal Astrocytic Hamartoma)
In the world of ocular oncology and retinal pathology, the diagnosis of a lesion such as a Retinal astrocytoma (also termed retinal astrocytic hamartoma, RA/RAH) demands both care and a clear understanding of its nature. These lesions arise from retinal glial (astrocytic) cells, often remain clinically silent, yet warrant recognition because of their associations and potential mimicry of more aggressive pathology.
Definition & Pathogenesis
Retinal astrocytoma (RA) is a benign glial tumour or hamartoma of the retina, originating from retinal astrocytes in the nerve fibre layer or inner retina.
In many cases, especially in the context of Tuberous Sclerosis Complex (TSC), the lesion is congenital or develops early; in other cases, it may be isolated (idiopathic) or associated with Neurofibromatosis type 1 (NF1).
When associated with TSC, the underlying mechanism involves mutation in TSC1 or TSC2 genes, leading to dysregulation of the mTOR pathway and hamartomatous proliferation of astrocytes.
Histopathologically, RAs are described as fibrous astrocytes with interlacing cytoplasmic processes, sometimes dystrophic calcifications, and in rare aggressive forms, pleomorphic cells, necrosis and mitoses.
Epidemiology & Associations
RAs are relatively rare as a discrete diagnosed entity whereas they are more common in patients with TSC: estimates vary, but approximately one-third to one-half of TSC patients will have retinal astrocytic hamartomas.
In non-TSC (idiopathic) cases, the lesion is often solitary, unilateral, and discovered incidentally at older age.
Associations beyond TSC include NF1, and more rarely, other retinal degenerations.
Clinical Features
Most patients are asymptomatic; RA is often an incidental finding on routine fundus examination.
Typical fundoscopic appearance:
A sessile or slightly elevated lesion in the nerve fibre layer, often near the optic disc, but it may occur anywhere in the retina (macula, mid-periphery, or periphery).
Variations in presentation:
- Unilateral or bilateral
- Solitary or multifocal
- Transparent/semitranslucent, or opaque/yellow-white
- Calcified or non-calcified
Rarely, the lesion may cause visual symptoms if located in the macula or optic disc, or if associated with exudation, retinal detachment, or neovascular glaucoma.
Typical Imaging & Investigation Findings
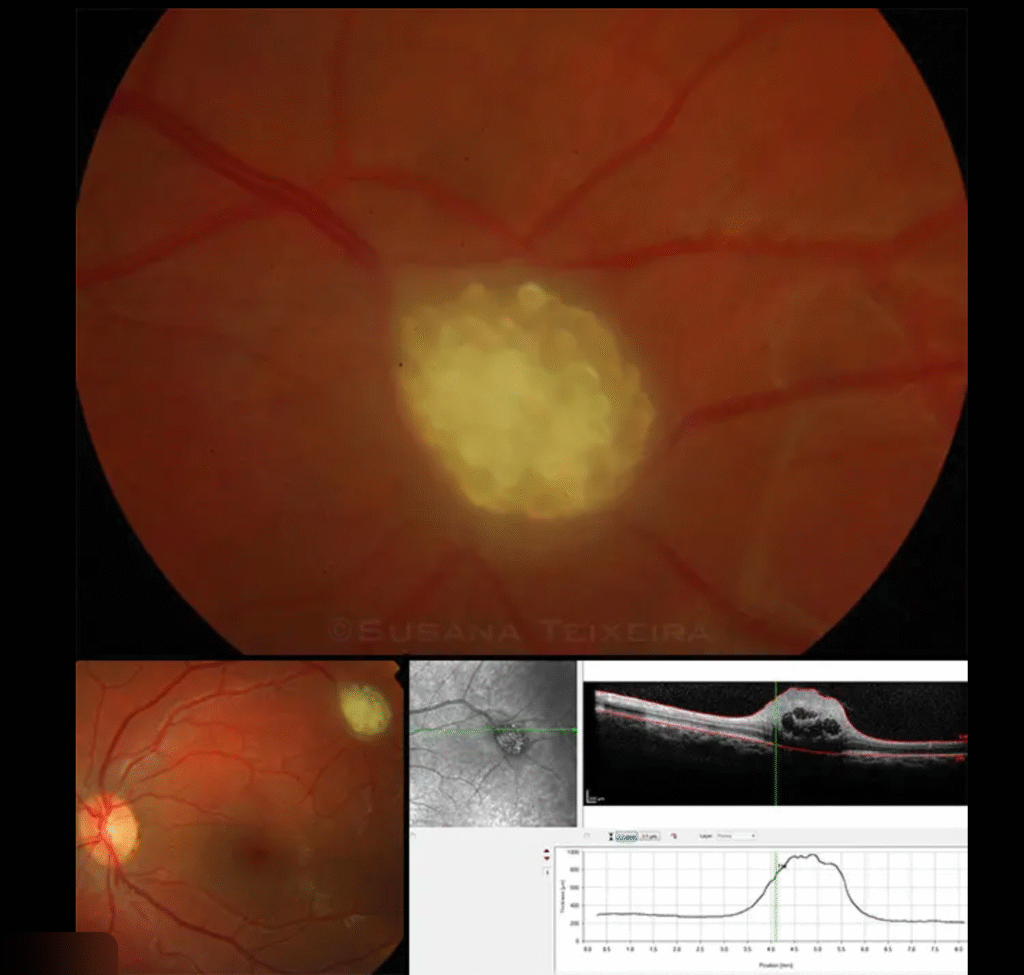
Image credit- AAO
Given the need to distinguish RA from more aggressive or malignant lesions, multimodal imaging is highly relevant.
1. Colour Fundus Photography
Documents the lesion’s colour, borders, topography, translucency or “mulberry” nodularity, and calcifications.
Serial imaging enables monitoring for change in size, colour, or exudation.
2. Fundus Autofluorescence (FAF)
Useful to assess metabolic activity of the retinal pigment epithelium and tumour interface; may show autofluorescent features if calcification or gliosis is present.
3. Spectral-Domain Optical Coherence Tomography (SD-OCT)
Cross-sectional visualization: RA typically lies in the inner retina/nerve fibre layer, sometimes with overlying vitreous protrusion, and may show hyper-reflectivity, shadowing if calcified, or possible intralesional cavities or cysts.
The transition between adjacent normal retina and tumour is often gradual.
Measurements of tumour thickness and retinal involvement can be taken with SD-OCT.
4. Ultrasonography (B-scan)
Useful for posterior lesions: assesses lesion thickness, internal reflectivity, posterior acoustic shadowing (often present in calcified lesions), and extension into vitreous or sub-retinal space.
5. Fluorescein Angiography and/or Indocyanine Green (ICG)
May show early surface vascularisation, delayed filling, or retained fluorescence; less frequently required in straightforward cases.
6. Systemic Work-Up (When Indicated)
Because of the strong association with TSC, when RA is identified—especially if bilateral, multifocal or in a young patient—a systemic evaluation (neurology, dermatology, genetics, renal/cardiac imaging) is warranted.
Differential Diagnosis
Recognition of RA is clinically important because the differential includes potentially aggressive lesions.
Key differentials include:
- Retinoblastoma: Particularly in children, this differential can be challenging when RA has a mulberry-like appearance. Features that favour RA include its inner retinal location, calcification with high reflectivity and shadowing, absence of dilated tortuous feeder vessels, and overall stability.
- Choroidal melanoma / amelanotic uveal melanoma: Usually arises from the choroid (rather than retina), often in older adults, may show outer-retinal involvement or “draping,” and lacks the characteristic calcification of RA.
- Choroidal osteoma: Can show calcification but is a choroidal lesion, often juxtapapillary, with a yellow-orange appearance rather than a glial retinal tumour.
- Other hamartomas or reactive glial proliferations: e.g., acquired retinal astrocytoma (more progressive) or reactive retinal astrocytic tumour.
Natural Course & Prognosis
In the majority of cases, RA are relatively quiescent lesions with little or no progression over years.
Many reports demonstrate long-term stability (for example, a seven-year follow-up of isolated RAH with no growth).
However, in a minority—especially in the TSC-associated group—RAs may demonstrate aggressive behaviour: rapid growth, exudation, retinal detachment, or neovascular glaucoma, sometimes necessitating enucleation.
Histologically, aggressive forms may show pleomorphism, necrosis, and extension beyond the retina.
No confirmed reports of metastasis from typical RA have been documented.
Management & Follow-Up
- Asymptomatic, stable lesions: Conservative management is appropriate — periodic monitoring with fundus photography and/or OCT, typically every 6–12 months (or more frequently if location is high-risk).
- Systemic evaluation: If the lesion is bilateral, multifocal, or occurs in a child, or there are other features suggestive of TSC/NF, referral for neurologic, dermatologic, and genetic assessment is indicated.
- Intervention: Rarely necessary, but may be considered in cases of:
- Vision-threatening complications (macular involvement, exudation, sub-retinal fluid)
- Secondary effects: retinal detachment, neovascular glaucoma
- Confirmed growth or alarming changes in imaging
Treatment options include photodynamic therapy (PDT), brachytherapy, vitrectomy, anti-VEGF therapy (for exudation), and mTOR inhibitors (in TSC-related cases). Evidence remains limited.
Clear documentation is vital: baseline imaging (fundus photo, OCT) and serial measurements are recommended so that any change in size, reflectivity, or new complications can be detected early.
Key Points
- Always consider RA in an asymptomatic yellow-white retinal lesion, especially in the inner retina or near the optic disc.
- Bilaterality or multifocality should prompt evaluation for TSC or NF1.
- Use multimodal imaging—fundus photography, OCT, ultrasound—for accurate characterisation.
- Serial imaging over time is more informative than a single exam.
- Misdiagnosis can lead to overtreatment; careful differentiation from malignant lesions is crucial.
Summary
Retinal astrocytoma is a rare but important entity in ophthalmology. While generally benign and stable, its potential associations (notably with Tuberous Sclerosis) and its ability to mimic more sinister pathology make it essential for clinicians to recognise its features, document it carefully, and monitor it over time. With proper assessment and follow-up, most patients with RA can be managed safely.
References
- EyeWiki. Retinal Astrocytic Hamartoma. American Academy of Ophthalmology. https://eyewiki.org/Retinal_Astrocytic_Hamartoma
- OUP Academic. Retinal Astrocytic Hamartoma: Diagnostic Challenges and Review.https://academic.oup.com/pmj/article/77/911/556/7039246
- JAMA Network. Shields CL, Eagle RC Jr, Shields JA. Retinal Astrocytic Hamartoma: Clinical Spectrum and Imaging Correlation. JAMA Ophthalmol. 2004;122(10):1522–1530.
- Nature Eye Journal. Multimodal Imaging in Retinal Astrocytic Hamartomas Associated with Tuberous Sclerosis Complex. 2022. https://www.nature.com/articles/s41433-022-02275-0
- PMC. Isolated Retinal Astrocytic Hamartoma: Long-term Follow-up and Imaging Features. 2023. https://pmc.ncbi.nlm.nih.gov/articles/PMC10476716/
- Lippincott Journals. Case of a Diagnostic Challenge: Retinal Astrocytic Hamartoma. Journal of Clinical Ophthalmology and Research. 2022.
- Singh AD, et al. Retinal Astrocytoma: Natural History and Management Strategies. Eye (Lond). 2005;19(6):651–657.