

Understanding Optic Nerve Glioma: A Pediatric Optic Pathway Tumor
Optic nerve glioma is a rare, typically slow-growing tumor that primarily affects children, with a median age of presentation around 6.5 years. Classified histologically as a pilocytic astrocytoma, this tumor originates from astrocytes—the supportive cells of the nervous system—and specifically involves the optic nerve. Though considered a low-grade tumor, its behavior can be highly variable, making management a complex and individualized decision.
Histopathology and Nature of the Tumor
Microscopically, optic nerve gliomas are composed of spindle-shaped pilocytic (hair-like) astrocytes interspersed with glial filaments. These characteristics are typical of pilocytic astrocytomas, which are WHO grade I tumors, suggesting a generally favorable prognosis in many cases. However, this does not guarantee a benign course. Some gliomas remain stable for years, while others can progress aggressively, extending intracranially and compromising vital structures.
Association with Neurofibromatosis Type I (NF1)
Approximately 30% of optic nerve glioma cases are associated with neurofibromatosis type I (NF1). This genetic disorder not only predisposes individuals to various benign and malignant tumors but also significantly influences the clinical course of optic nerve glioma. Interestingly, in NF1 patients, the tumors often exhibit a more indolent course, and spontaneous regression has occasionally been reported. Therefore, the prognosis in NF1-associated optic glioma is generally more favorable compared to sporadic cases.
Clinical Presentation
Symptoms:
- Progressive visual loss is typically the earliest and most common complaint.
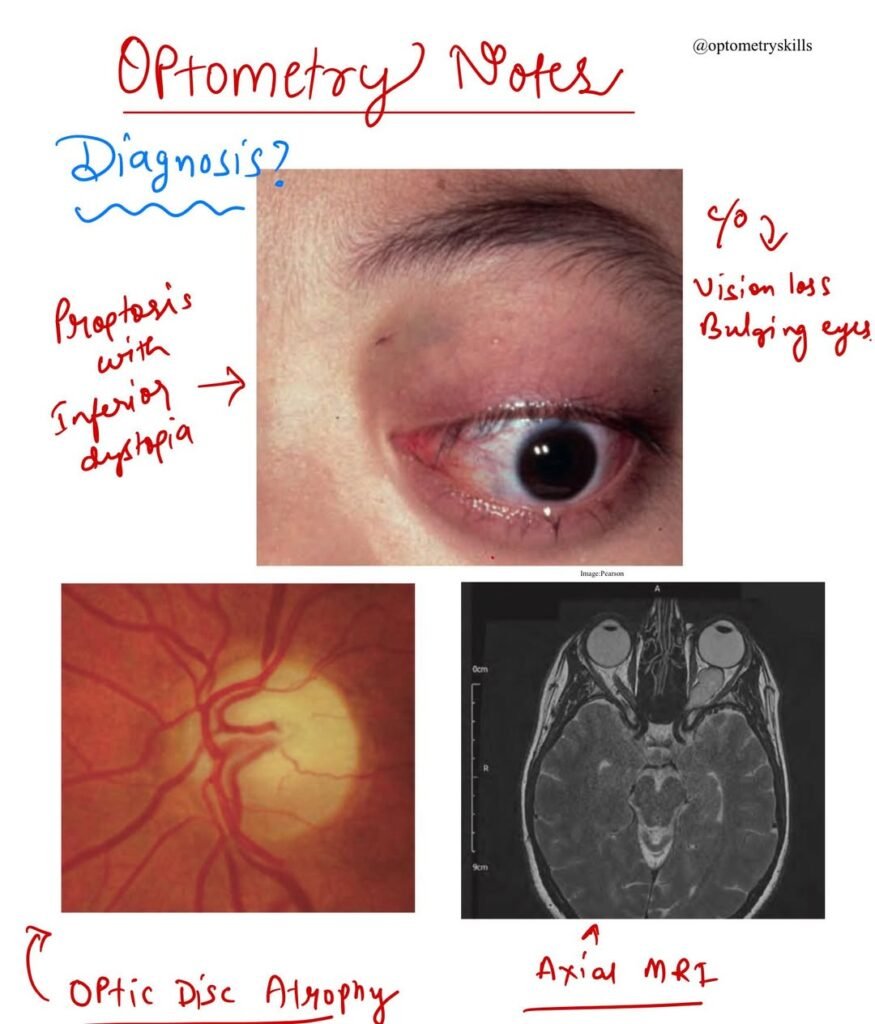
- Proptosis (bulging of the eye) follows as the tumor enlarges, although in some cases, it may precede visual symptoms.
- Rarely, an acute visual decline may result from hemorrhage within the tumor.
Signs:
- Non-axial proptosis is commonly observed, often with temporal or inferior globe displacement.
- The optic nerve head may initially appear swollen but tends to become atrophic over time.
- Opticociliary collaterals, central retinal vein occlusion, and other fundus abnormalities may be present.
- Intracranial extension to the optic chiasm and hypothalamus is a serious complication that can develop in progressive cases.
Diagnostic Investigations
- MRI is the imaging modality of choice, providing detailed visualization of the optic nerve and any intracranial extension.
- CT scans, especially in NF1 patients, typically show a fusiform enlargement of the optic nerve with distinct borders due to the intact dural sheath. In contrast, non-NF1 cases often exhibit a more irregular nerve contourwith hypodense regions.
Management Strategies
Treatment of optic nerve glioma is tailored according to tumor size, location, progression, visual function, and cosmetic concerns. Given the tumor’s intrinsic position within the optic nerve, complete surgical resection leads to irreversible vision loss in the affected eye, a major consideration in therapeutic planning.
- Observation
In cases where the glioma appears stable on imaging, especially in NF1 patients, observation with serial MRI scans may be appropriate. This is particularly valid when vision is preserved and there are no significant cosmetic or neurological issues. - Surgical Excision
Indicated in patients with progressive growth, poor vision, or significant proptosis. The primary objective is to prevent chiasmal involvement. Depending on the tumor’s extent, an intracranial surgical approach may be necessary for optimal removal. - Radiotherapy and Chemotherapy
These are considered in non-resectable tumors, especially when there is extensive intracranial extension. The combination may help to control tumor growth and preserve neurological function, though risks of long-term radiation side effects in children must be carefully weighed.
Malignant Transformation and Rare Variants
Malignant glioma (glioblastoma) of the optic nerve is exceedingly rare, tends to occur in adult males, and carries a poor prognosis. Unlike pilocytic astrocytomas, glioblastomas are aggressive, fast-growing, and often resistant to conventional therapy.
Conclusion
Optic nerve glioma remains a unique challenge in pediatric neuro-ophthalmology. While often associated with a benign course—especially in NF1 cases—the potential for vision loss, disfigurement, and intracranial involvement makes early diagnosis and personalized treatment essential. A multidisciplinary approach, involving ophthalmologists, neurologists, oncologists, and neurosurgeons, is key to achieving the best outcomes for affected children.