
Choroidal Melanoma: A Clinically Oriented Overview
Choroidal melanoma is the most common primary intraocular malignancy in adults and constitutes the majority of uveal melanomas. Although relatively rare in the general population, it carries significant clinical importance due to its potential for visual morbidity and life-threatening metastasis. The tumor arises from melanocytes within the choroid and typically presents in late middle age, with a peak incidence around 60 years.
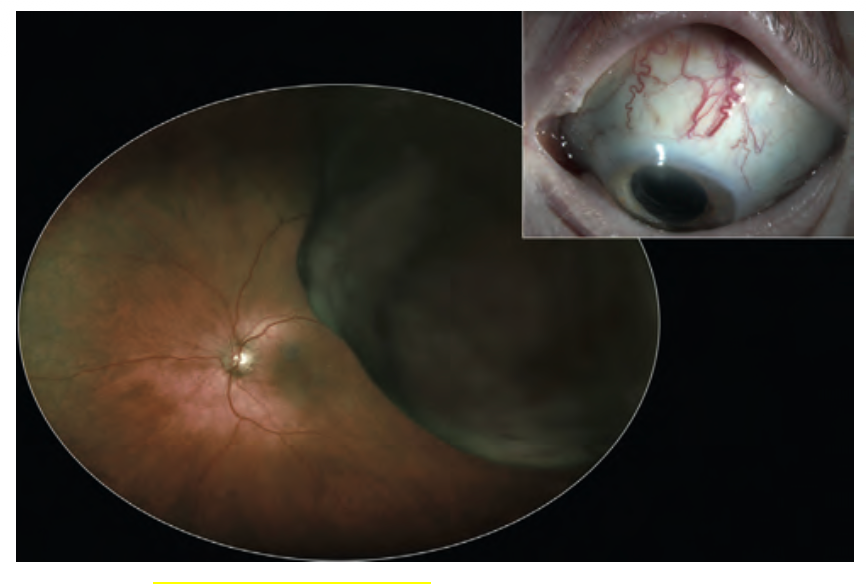
Choroidal Melanoma- Image courtesy- C berry, Kanki
Epidemiology and Etiology
Choroidal melanoma accounts for approximately 80–90% of uveal melanomas. The incidence is estimated at around 5 per million population annually. Most cases occur sporadically, and unlike cutaneous melanoma, these tumors are not strongly associated with ultraviolet exposure. Instead, they exhibit distinct molecular characteristics, including mutations in genes such as GNAQ, GNA11, and BAP1.
Risk factors include fair skin, light-colored irides, pre-existing choroidal nevi, and ocular melanocytosis. Genetic predisposition plays an important role, particularly in determining metastatic potential.
Pathology and Growth Pattern
The tumor originates from melanocytes in the choroidal stroma and may consist of:
- Spindle cells – elongated, less aggressive, better prognosis
- Epithelioid cells – pleomorphic, mitotically active, associated with poorer prognosis
- Mixed cell type – most common
A hallmark feature of choroidal melanoma is its ability to penetrate Bruch’s membrane. When this occurs, the tumor expands into the subretinal space, forming a mushroom-shaped or collar-button configuration, which is highly suggestive of malignancy.
Tumor extension may occur through scleral emissary channels and vortex veins, leading to extrascleral spread and increasing the risk of systemic dissemination.
Clinical Features
Symptoms
Many patients are asymptomatic in early stages, and lesions are often detected incidentally during routine examination. When symptomatic, patients may present with:
- Blurred or reduced vision
- Visual field defects
- Photopsia (flashes of light)
- Floaters
Symptoms are usually related to tumor size, location, and associated retinal changes.
Signs
Fundoscopic examination reveals characteristic findings:
- Solitary, elevated dome-shaped mass
- Gray-brown pigmentation, though amelanotic variants may occur
- Irregular margins
- Overlying orange pigment (lipofuscin)
- Subretinal fluid, sometimes leading to exudative retinal detachment
- Associated hemorrhage may be present
Approximately 20% of tumors break through Bruch’s membrane, producing the classic mushroom or collar-stud appearance.
Additional clinical signs may include:
- Sentinel vessels
- Choroidal folds
- Secondary glaucoma
- Rubeosis iridis
- Cataract in advanced disease
Differential Diagnosis
Differentiation from benign and other malignant lesions is critical.
Pigmented Lesions
- Choroidal nevus – flat or minimally elevated, often with drusen and absence of subretinal fluid
- Melanocytoma – deeply pigmented lesion, commonly at the optic disc
- CHRPE – flat, well-defined lesion with lacunae
- Choroidal hemorrhage
Non-pigmented Lesions
- Choroidal hemangioma – orange-red, smooth, dome-shaped lesion
- Metastatic tumors – often associated with extensive retinal detachment
- Granulomas (e.g., tuberculosis, sarcoidosis)
- Posterior scleritis – typically painful
- Choroidal neovascular lesions
Careful clinical and imaging evaluation is necessary to distinguish these entities.
Investigations
Diagnosis is often clinical but supported by imaging:
Ultrasonography (B-scan)
- Low to medium internal reflectivity
- Acoustic hollowing
- Choroidal excavation
- Mushroom configuration when Bruch’s membrane is breached
Optical Coherence Tomography (OCT)
- Detects subretinal fluid and retinal changes
Fundus Fluorescein Angiography (FFA)
- Shows intrinsic tumor circulation and late leakage
Fundus Autofluorescence (FAF)
- Highlights lipofuscin as hyperautofluorescence
Indocyanine Green Angiography (ICG)
- Better delineation of tumor extent
MRI
- Useful for detecting extrascleral extension
Biopsy and Genetic Testing
- Reserved for uncertain cases
- Genetic profiling (e.g., BAP1 mutation) helps assess metastatic risk
Metastasis and Prognosis
Choroidal melanoma spreads hematogenously, most commonly to the liver, followed by the lungs and bones. At presentation, only a small percentage of patients have detectable metastasis, but long-term risk remains high.
Mortality rates approach 50% at 10 years.
Poor Prognostic Indicators
- Large tumor size
- Epithelioid cell predominance
- High mitotic activity
- Extrascleral extension
- High-risk genetic profile (e.g., BAP1 mutation)
Management
Management is individualized, aiming to preserve the eye and vision while controlling tumor growth.
Globe-Preserving Treatments
Brachytherapy (Plaque Radiotherapy)
- Standard treatment for small to medium tumors
- Radioactive plaque sutured to sclera
- Gradual tumor regression over months to years
- Complications include radiation retinopathy and optic neuropathy
Proton Beam Radiotherapy
- Used for larger or posterior tumors
- Precise delivery of radiation
Stereotactic Radiotherapy
- Focused radiation beams
- Effective but associated with higher complication rates
Transpupillary Thermotherapy (TTT)
- Infrared laser-induced hyperthermia
- Used for small tumors or adjunct therapy
Surgical Management
Local Resection (Choroidectomy)
- Selected cases
Enucleation
- Indicated for large tumors, painful blind eye, or extensive involvement
- Ensures complete removal when vision cannot be preserved
Emerging Therapies
Newer modalities such as light-activated targeted therapies (e.g., AU-011) are under investigation, particularly for small melanomas, offering promising future treatment options.
Systemic Evaluation
Given the high risk of liver metastasis, systemic surveillance is essential:
- Liver function tests
- Liver imaging (ultrasound, MRI)
- Additional imaging when indicated
Conclusion
Choroidal melanoma is a serious intraocular malignancy with significant implications for both vision and survival. Its characteristic clinical appearance—an elevated pigmented lesion with possible mushroom configuration, orange pigment, and subretinal fluid—provides key diagnostic clues.
Early detection, accurate differentiation from benign lesions, and appropriate imaging are essential for optimal management. Advances in radiotherapy and genetic profiling have improved local tumor control, but metastatic disease remains the principal determinant of prognosis.
A comprehensive and multidisciplinary approach is essential to ensure timely diagnosis, effective treatment, and long-term monitoring.